October 2011—The Editors are re-posting this expert Commentary by Dr. Weber on the heels of FDA’s August 2011 warning that correcting pelvic organ prolapse (POP) with transvaginal placement of mesh may be riskier than more traditional surgical approaches—without offering greater benefit. To read about the FDA’s warning, click here.
The author reports no financial relationships relevant to this article.
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries1,2 and visit http://www.fda.gov/cdrh/devadvice/314.html.
In a nutshell, the 510(k) process requires only that an applicant demonstrate that a new medical device has “substantial equivalence” to an already legally marketed device, known as the predicate, which may also have been cleared only through the 510(k) process. That means it’s possible to have generations of products cleared on the basis of one predicate device that was itself never studied adequately.
Indeed, this is the case with most medical devices that have been sold for the surgical treatment of POP and UI—from before the ProteGen Sling (Boston Scientific), through Tension-Free Vaginal Tape (TVT) (Gynecare), and continuing with the newest devices.
The story of the ProteGen Sling (FIGURE) offers a cautionary tale about what can go wrong when new devices are cleared by the FDA through 510(k), rather than evaluated through rigorous clinical trials, as drugs are. More recently, experience with the ObTape (Mentor Corporation) followed virtually the same trajectory of events; the product was pulled from the market in 2006 and is now the focus of lawsuits nationwide.
Fortunately, for our patients, experience with TVT (Gynecare) has been favorable. Although TVT was also cleared by the FDA through 510(k), clinical research performed after TVT was introduced has demonstrated its effectiveness and relative safety. Indeed, TVT has revolutionized the treatment of stress UI in women—and, even, our understanding of its etiology.
Several companies are capitalizing on the success of TVT by introducing competing products that are designed to be 1) similar enough to ride on the coattails of TVT yet 2) different enough to capture their own share of market—without evidence of safety or effectiveness required. Even Gynecare (part of Ethicon Women’s Health and Urology, a subsidiary of Johnson & Johnson) has introduced TVT SECUR to compete with its own TVT—again, without independent evidence of safety or effectiveness.
The current market in devices for stress UI is a moving target that makes it nearly impossible—even for research organizations, such as the federally funded Pelvic Floor Disorders Network, that are independent of industry—to develop and implement sound clinical trials of those devices. Why do I say “moving target”? First, there are no assurances that any device chosen for study will remain the same for the duration of a trial. Second, there is no way to foresee which products will be abandoned over the time required for a large clinical trial.
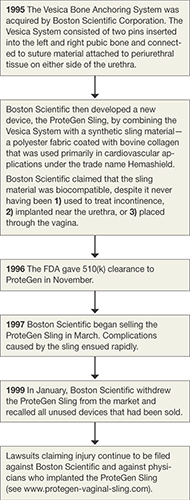
FIGURE The saga of the ProteGen Sling
Transparency over what might be considered “experimental”
Until the FDA changes its process—to one in which 1) medical devices are adequately assessed before they reach market and 2) postmarketing surveillance is required—it’s our duty to insist on evidence of safety and effectiveness before adopting the latest and greatest products that companies have to offer.
Of all the questions that a patient might ask before treatment, three of the most important, surely, are: